Image qu’on te donne une liste de 100 acides aminés qui forme une protéine. Si avec un algorithme, nous devions trouver la vraie structure de celle-ci avec le moins d’énergie parmi toutes les structures possibles, nous prendrions plus longtemps que le temps de l’univers ! Et cela même si on arrive à tester chaque configuration à seulement une nanoseconde ! Pourtant, la protéine se met spontanément sous cette forme en quelques millisecondes ou même microsecondes. Voilà le paradoxe de Levinthal.
Avant de rentrer dans le vif du sujet, commençons par reconnaître l’importance de la structure d’une protéine. Les protéines sont le fondement de tous les organismes et donc de la vie sur la terre. En effet, ce sont eux qui travaillent dans les cellules, transmettre des signaux pour réguler les tissus et les organes du corps, et bien plus. C’est donc la base et connaitre les formes que les protéines prennent, permettrait des avancements médicaux astronomiques.
Mais, comme expliqué auparavant, cela s’avère d’une difficulté énorme. Cependant, assez récemment, le jeu a changé. En effet, DeepMind, une subsidiaire du géant Alphabet (aka Google) à développé grâce à de la “machine learning” un programme nommé AlphaFold qui vise justement à utiliser les dernières techniques pour faire des prédictions de structure de protéines.

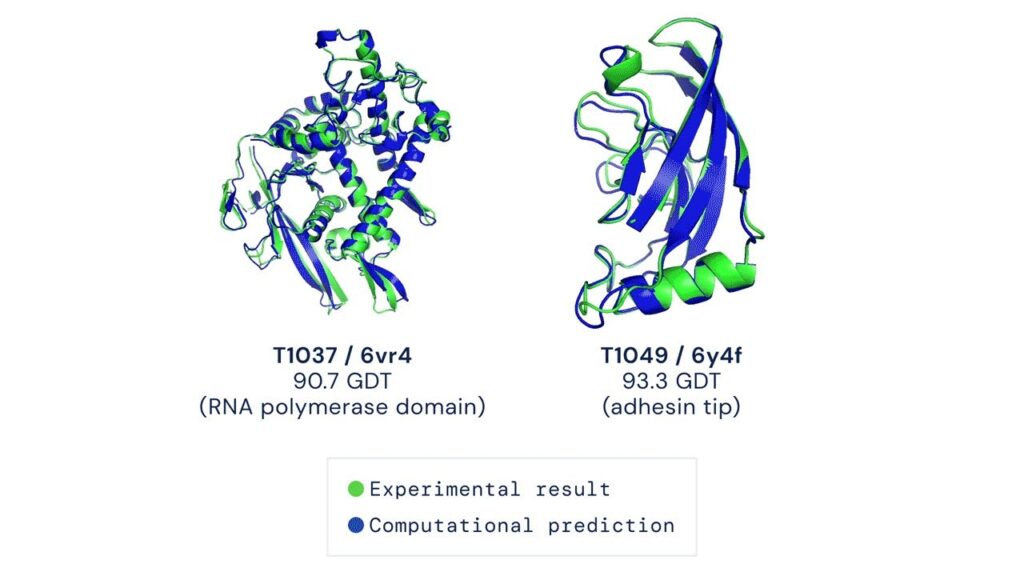
Leur première version est sortie en 2018, a été classé premier au classement général de la 13ᵉ Évaluation critique de la prédiction des structures (CASP), une compétition qui s’organise tous les deux ans et qui vise a classé les programmes de prédictions. Leur 2ᵉ version (qui est très différente à la 1ʳᵉ) a refait cet exploit en 2020 avec un score de 90 et une précision beaucoup plus haute que tous les autres groupes. Pour la compétition, les comparaisons se font avec des structures déterminées par une expérience en laboratoire par des techniques comme la cristallographie aux rayons X, cryomicroscopie électronique et résonance magnétique nucléaire. Comme les noms laissent imaginer, ces techniques sont chères et consomment du temps. Les résultats de AlphaFold sont dits aussi précis que les méthodes expérimentales et ont donc été lourdement applaudi dans le monde scientifique pour ces avancements technologiques.
Le nouveau programme utilise des sous-réseaux interconnectés dans un modèle unifié basé sur la reconnaissance de formes. Oui, pas facile à comprendre. Mais en quelques termes plus simples, les modules affinent progressivement les relations entre les résidus d’acides aminés et les alignements de séquences. Le processus de raffinement, qui ressemble à l’assemblage d’un puzzle, se met à jour de manière itérative et filtre à chaque fois les données pertinentes.
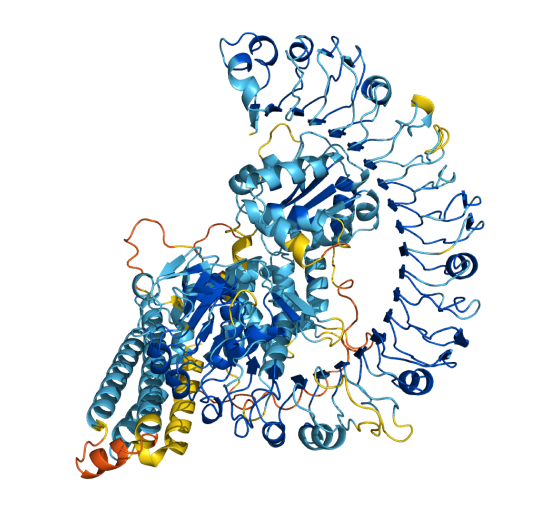
En opposition a son prédécesseur, la physique locale est appliquée uniquement comme étape de raffinement final, en ajustant légèrement la structure prédite.
La compagnie a maintenant prédit les structures de 200 millions de protéines — presque tous ceux connus dans le monde. De plus, ils ont ouvert leurs résultats au public qui peut rechercher la protéine qu’il veut dans la base de donnée. “Cela change la donne”, déclare Andrei Lupas, biologiste évolutionniste à l’Institut Max Planck de biologie du développement à Tübingen, en Allemagne. Et d’autre affirme que, même si le système n’est pas parfait, le problème des structures de protéines est en quelque sorte résolue. Il n’y donc aucun doute que cette technologie a permis une étape révolutionnaire dans la compréhension de la biologie.
Jean-Luc pour le KapTech 🔥